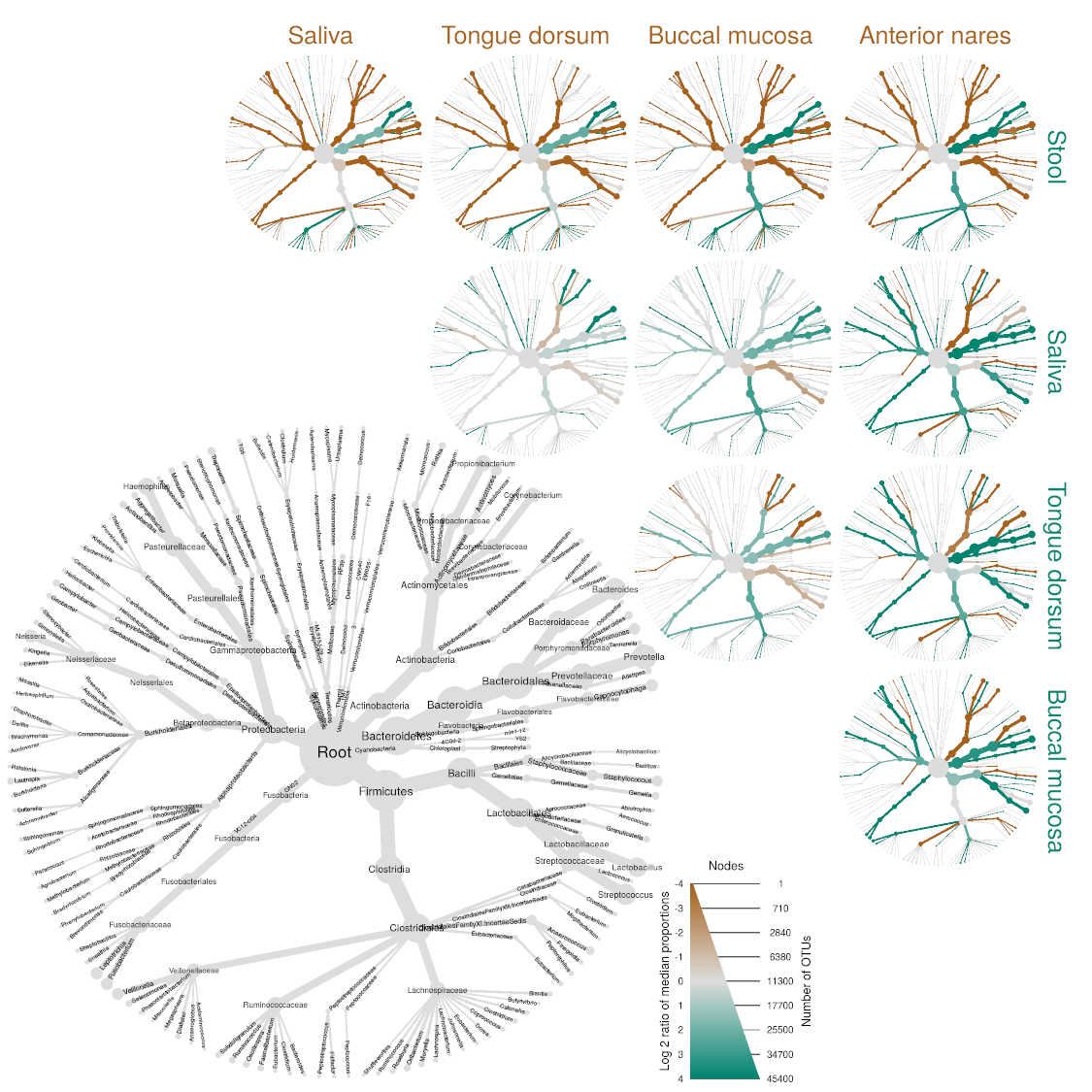
Metacoder is an R package for reading, plotting, and manipulating
large taxonomic data sets, like those generated from modern
high-throughput sequencing, like metabarcoding (i.e. amplification
metagenomics, 16S metagenomics, etc). It provides a tree-based
visualization called “heat trees” used to depict statistics for every
taxon in a taxonomy using color and size. It also provides various
functions to do common tasks in microbiome bioinformatics on data in the
taxmap format defined by the taxa package,
such as:
vegantaxa
package.phyloseq format and the
taxa formatThis project is available on CRAN and can be installed like so:
install.packages("metacoder")You can also install the development version for the newest features, bugs, and bug fixes:
install.packages("devtools")
devtools::install_github("grunwaldlab/metacoder")All the documentation for metacoder can be found on our
website here:
https://grunwaldlab.github.io/metacoder_documentation/
The function that simulates PCR requires primersearch
from the EMBOSS tool kit to be installed. This is not an R package, so
it is not automatically installed. Type ?primersearch after
installing and loading metacoder for installation instructions.
Many of these operations can be done using other packages like
phyloseq, which also provides tools for diversity analysis.
The main strength of metacoder is that its functions use
the flexible data types defined by taxa, which has powerful
parsing and subsetting abilities that take into account the hierarchical
relationship between taxa and user-defined data. In general,
metacoder and taxa are more of an abstracted
tool kit, whereas phyloseq has more specialized functions
for community diversity data, but they both can do similar things. I
encourage you to try both to see which fits your needs and style best.
You can also combine the two in a single analysis by converting between
the two data types when needed.
If you use metcoder in a publication, please cite our article in PLOS Computational Biology:
Foster ZSL, Sharpton TJ, Grünwald NJ (2017) Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLOS Computational Biology 13(2): e1005404. https://doi.org/10.1371/journal.pcbi.1005404
Metacoder is under active development and many new features are planned. Some improvements that are being explored include:
To see the details of what is being worked on, check out the issues tab of the Metacoder Github site.
This work is subject to the MIT License.
Metacoder’s major dependencies are taxa,
taxize, vegan, igraph,
dplyr, and ggplot2.
This package includes code from the R package ggrepel to handle label
overlap avoidance with permission from the author of ggrepel Kamil Slowikowski. We included the
code instead of depending on ggrepel because we are using
functions internal to ggrepel that might change in the
future. We thank Kamil Slowikowski for letting us use his code and would
like to acknowledge his implementation of the label overlap avoidance
used in metacoder.
We would like to hear about users’ thoughts on the package and any errors they run into. Please report errors, questions or suggestions on the issues tab of the Metacoder Github site. We also welcome contributions via a Github pull request. You can also talk with us using our Google groups site.